Benchmarking {epistack}
Foreword
We recently developed an R package, named {epistack}, to visualise
epigenomic data. Epistack is available on
github and
Bioconductor.
The best place to learn more about it is probably the package’s vignette.
The following is a silly attempt at benchmarking {epistack} againts the
great {EnrichedHeatmap}.
TLDR: both are fast enough.
Generating benchmark datasets
To compare {epistack} with other methods, we will use an artificially generated
dataset. We will generate artificial datasets of various regon length using
a function defined bellow (there is no need to understand how this function
works, it’s simply generate a dumy object that can be input to
plotEpistack()).
library(GenomicRanges)
library(epistack)
epidataset <- function(n) {
# fist we generate a fake epigenetic signal matrix
mat = matrix(nrow = n, ncol = 50)
for(i in seq_len(nrow(mat))) {
mat[i, ] = runif(50) +
c(sort(abs(rnorm(50)))[1:25], rev(sort(abs(rnorm(50)))[1:25]))*i/(n/4)
}
# we then embeded the matrix in a GRanges object
testdat <- GRanges(
rep("chr1", n),
IRanges(rep(1, n), rep(2, n)),
mcols = mat
)
testdat$expr <- exp(seq(from = 0, to = 3, length.out = n))
testdat <- testdat[seq(length(testdat), 1),]
testdat <- addBins(testdat, nbins = 5)
testdat
}
The function is working as intended:
epidataset(100)[, 49:52]
#> GRanges object with 100 ranges and 4 metadata columns:
#> seqnames ranges strand | mcols.V49 mcols.V50 expr bin
#> <Rle> <IRanges> <Rle> | <numeric> <numeric> <numeric> <numeric>
#> [1] chr1 1-2 * | 0.952068 0.179671 20.0855 1
#> [2] chr1 1-2 * | 0.478568 0.340969 19.4860 1
#> [3] chr1 1-2 * | 0.412846 0.497083 18.9044 1
#> [4] chr1 1-2 * | 0.684431 0.437699 18.3401 1
#> [5] chr1 1-2 * | 0.580179 0.901338 17.7927 1
#> ... ... ... ... . ... ... ... ...
#> [96] chr1 1-2 * | 0.0516165 0.7985905 1.12886 5
#> [97] chr1 1-2 * | 0.2357467 0.3174062 1.09517 5
#> [98] chr1 1-2 * | 0.6359180 0.2045663 1.06248 5
#> [99] chr1 1-2 * | 0.6136850 0.0915456 1.03077 5
#> [100] chr1 1-2 * | 0.0981223 0.7333909 1.00000 5
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
epidataset(10000)[, 49:52]
#> GRanges object with 10000 ranges and 4 metadata columns:
#> seqnames ranges strand | mcols.V49 mcols.V50 expr bin
#> <Rle> <IRanges> <Rle> | <numeric> <numeric> <numeric> <numeric>
#> [1] chr1 1-2 * | 0.890472 0.695199 20.0855 1
#> [2] chr1 1-2 * | 0.744390 0.424815 20.0795 1
#> [3] chr1 1-2 * | 0.443652 0.298934 20.0735 1
#> [4] chr1 1-2 * | 0.954323 0.983730 20.0675 1
#> [5] chr1 1-2 * | 0.307858 0.894837 20.0614 1
#> ... ... ... ... . ... ... ... ...
#> [9996] chr1 1-2 * | 0.342960 0.6116082 1.0012 5
#> [9997] chr1 1-2 * | 0.389621 0.0192827 1.0009 5
#> [9998] chr1 1-2 * | 0.499694 0.0725932 1.0006 5
#> [9999] chr1 1-2 * | 0.886972 0.9410834 1.0003 5
#> [10000] chr1 1-2 * | 0.243264 0.0821325 1.0000 5
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
The datasets can then be plotted with epistack:
plotEpistack(
epidataset(10000),
zlim = c(0, 3), ylim = c(0, 4), tints = "dodgerblue",
patterns = "mcols", titles = "Mark level", x_labels = c("", "middle", ""),
metric_col = "expr", metric_title = "Expression"
)
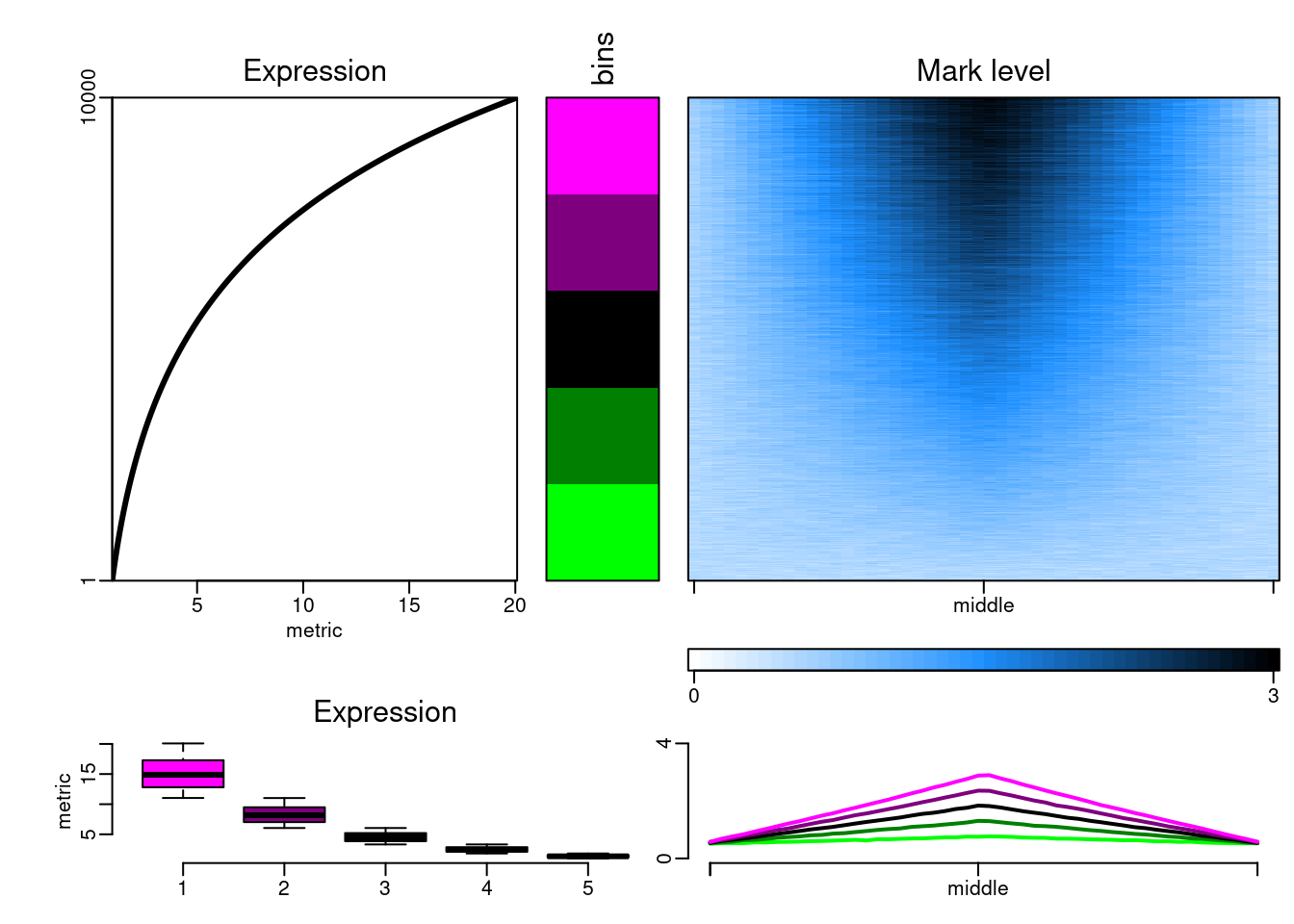
We can now generate a benchmark dataset containing GRanges objects of various lengths.
sizes <- c(
1000, 2000, 5000,
10000, 20000, 50000,
100000, 200000
)
benchdata <- lapply(sizes, epidataset)
Benchmarking epistack()
runtimes <- sapply(benchdata, function(x) system.time(
plotEpistack(
x,
zlim = c(0, 3), ylim = c(0, 4), tints = "dodgerblue",
patterns = "mcols", titles = "Mark level", x_labels = c("", "middle", ""),
metric_col = "expr", metric_title = "Expression"
)
)["elapsed"])
runtimes2 <- sapply(benchdata, function(x) system.time(
plotEpistack(
x,
zlim = c(0, 3), ylim = c(0, 4), tints = "dodgerblue",
patterns = "mcols", titles = "Mark level", x_labels = c("", "middle", ""),
metric_col = "expr", metric_title = "Expression"
)
)["elapsed"])
library(ggplot2)
data.frame(size = sizes, time = runtimes) |>
ggplot(aes(x = size, y = time)) +
geom_line() +
geom_point(shape = 3) +
theme_bw(base_size = 14) +
labs(x = "Number of regions", y = "Time (s)",
title = "plotEpistack() and EnrichedHeatmap() benchmark") +
coord_cartesian(ylim = c(0, 15))
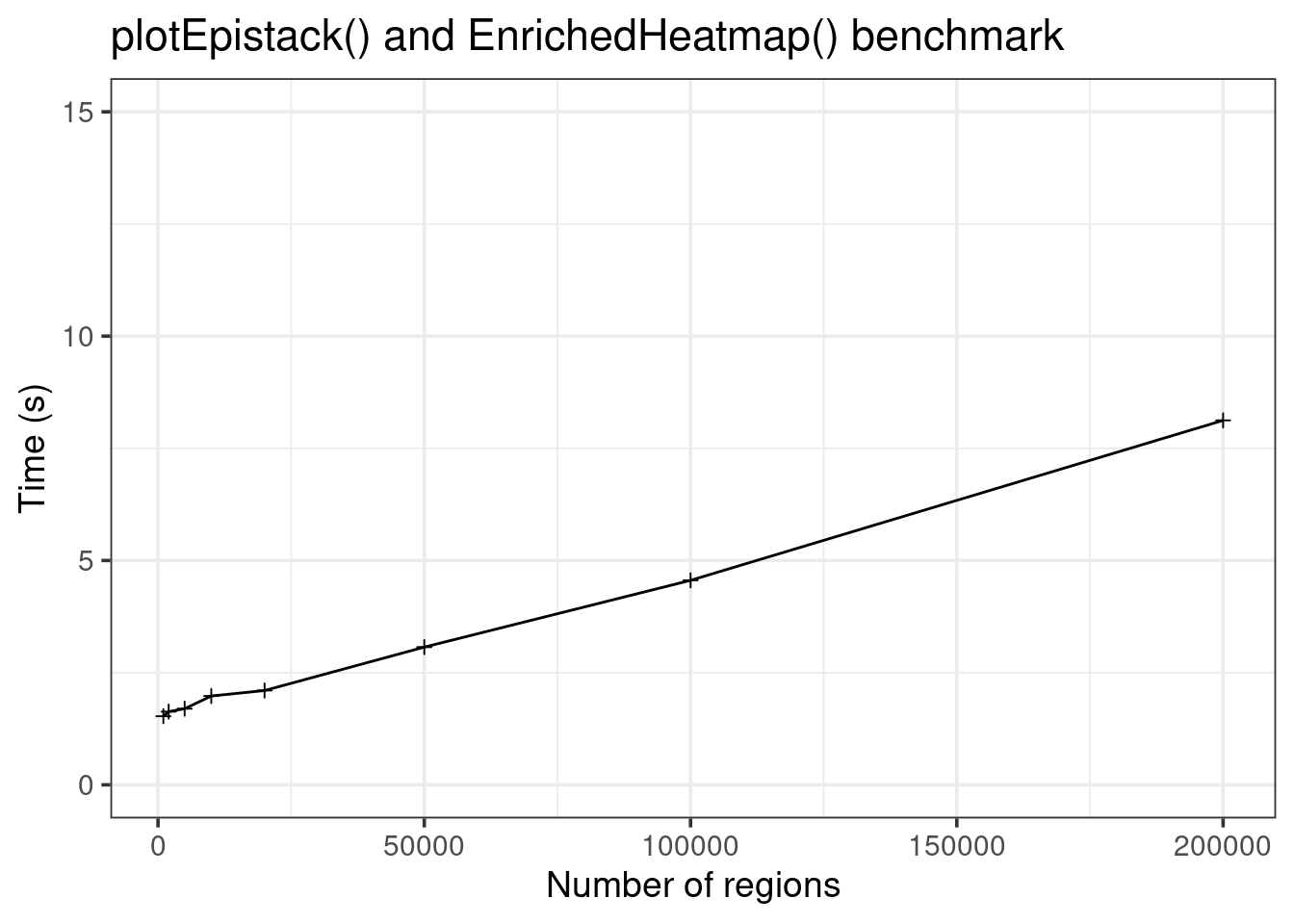
Comparison with EnrichedHeatmap
First we transform the benchmark dataset into the format needed by {EnrichedHeatmap}:
library(EnrichedHeatmap)
# we first make a dummy normalizedMatrix object to later steal its attributes
load(
system.file("extdata", "chr21_test_data.RData", package = "EnrichedHeatmap")
)
ehmat <- normalizeToMatrix(
meth,
resize(cgi, width = 1, fix = "center"),
value_column = "meth", mean_mode = "absolute",
extend = 2500, w = 100, smooth = TRUE
)
# we then apply the attributes to the signal matrices in benchdata
ehbenchdata <- lapply(
benchdata,
function(x) {
mat <- as.matrix(mcols(x)[, 1:50])
mdim <- dim(mat)
mdimnames <- dimnames(mat)
mostattributes(mat) <- attributes(ehmat)
dim(mat) <- mdim
dimnames(mat) <- mdimnames
mat
}
)
We can now call EnrichedHeatmap:
col_fun = circlize::colorRamp2(c(0, 1.5, 3), c("white", "dodgerblue", "black"))
EnrichedHeatmap(
ehbenchdata[[1]], col = col_fun,
name= "Mark level", column_title = "Mark level")
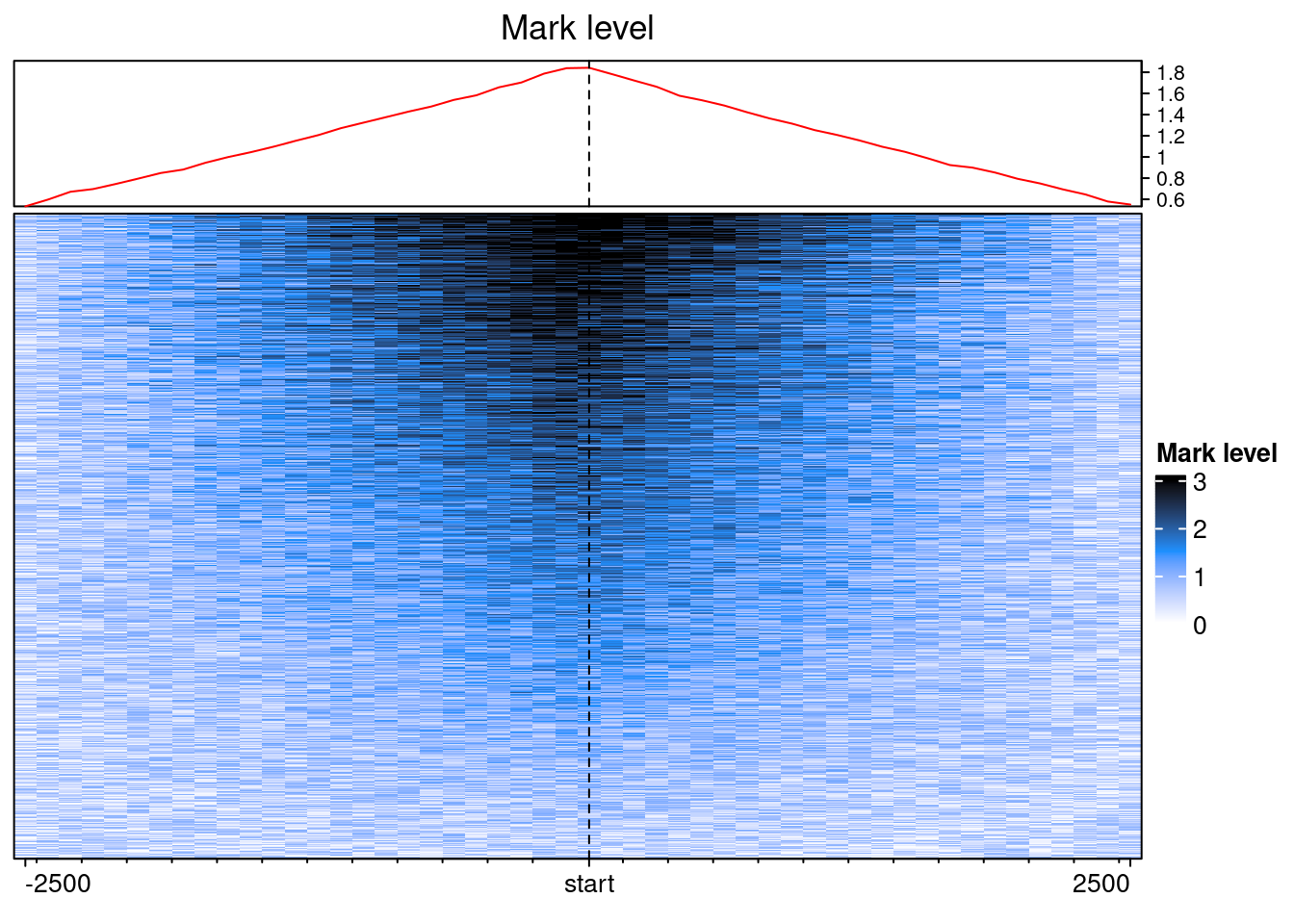
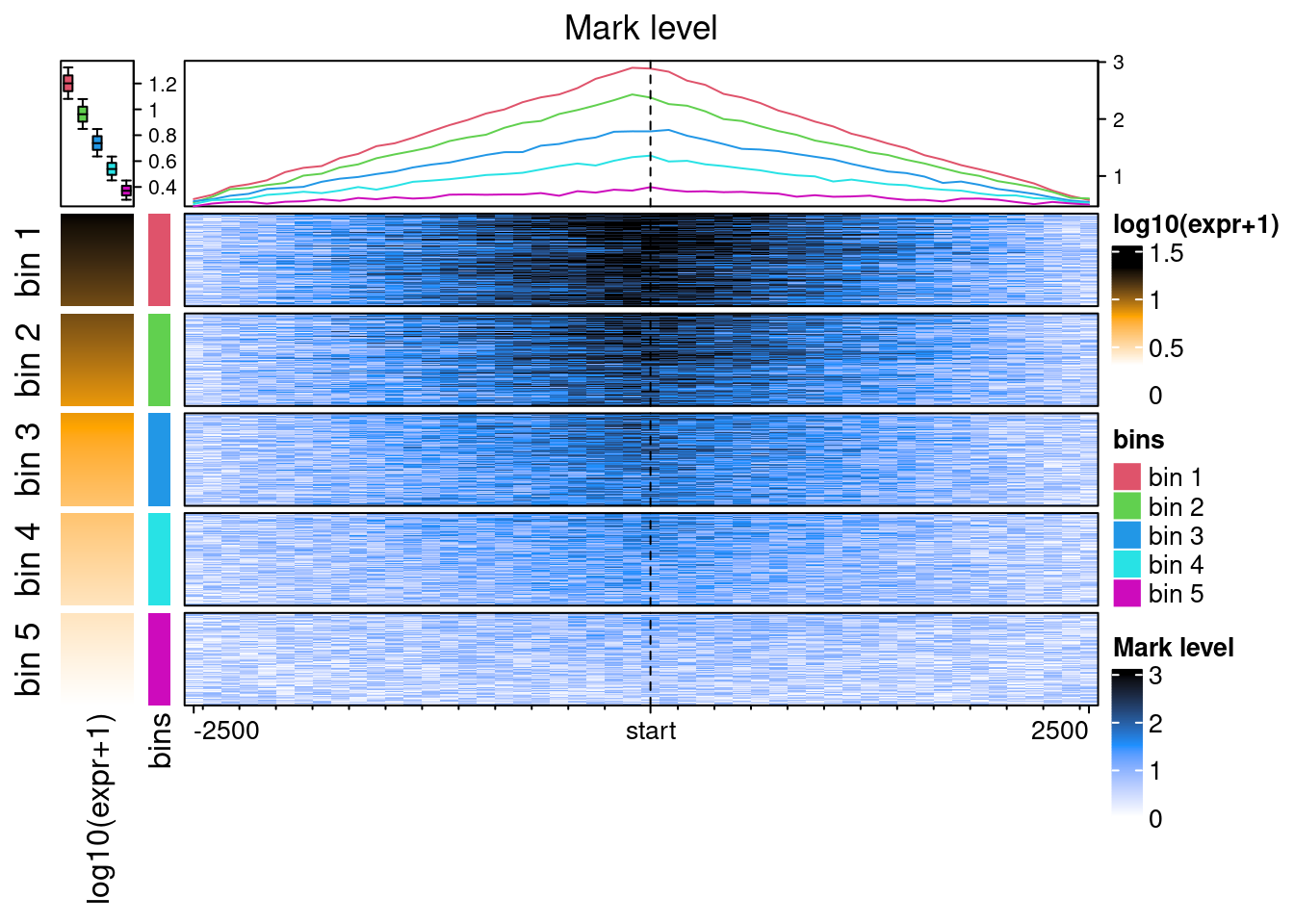
With some fiddeling, it is possible to make EnrichedHeatmap outputs equivalent to the plotEpistack’s outputs.
ht_list <- Heatmap(
log10(benchdata[[1]]$expr+1),
col = c("white", "orange", "black"), name = "log10(expr+1)",
show_row_names = FALSE, width = unit(10, "mm"),
top_annotation = HeatmapAnnotation(
summary = anno_summary(
gp = gpar(fill = 2:6),
outline = FALSE, axis_param = list(side = "right")
)
)
) +
Heatmap(
paste("bin", benchdata[[1]]$bin),
col = structure(2:6, names = paste("bin", 1:5)),
name = "bins",
show_row_names = FALSE, width = unit(3, "mm")
) +
EnrichedHeatmap(
ehbenchdata[[1]], col = col_fun,
name= "Mark level", column_title = "Mark level",
top_annotation = HeatmapAnnotation(
lines = anno_enriched(gp = gpar(col = 2:6))
)
)
draw(
ht_list,
split = paste("bin", benchdata[[1]]$bin), cluster_rows = FALSE
)
We can now run a benchmark. Note that there is currently a bug in EnrichedHeatmap preventing us to generate the Heatmaps with more than 100.000 regions unless we shut off rasterization.
runtimes2 <- sapply(1:8, function(i) system.time({
ht_list <- Heatmap(
log10(benchdata[[i]]$expr+1),
col = c("white", "orange", "black"), name = "log10(expr+1)",
show_row_names = FALSE, width = unit(10, "mm"),
top_annotation = HeatmapAnnotation(
summary = anno_summary(
gp = gpar(fill = 2:6),
outline = FALSE, axis_param = list(side = "right")
)
)
) +
Heatmap(
paste("bin", benchdata[[i]]$bin),
col = structure(2:6, names = paste("bin", 1:5)),
name = "bins",
show_row_names = FALSE, width = unit(3, "mm")
) +
EnrichedHeatmap(
ehbenchdata[[i]], col = col_fun,
name= "Mark level", column_title = "Mark level",
top_annotation = HeatmapAnnotation(
lines = anno_enriched(gp = gpar(col = 2:6))
)
)
draw(
ht_list,
split = paste("bin", benchdata[[i]]$bin), cluster_rows = FALSE,
use_raster = FALSE
)
})["elapsed"])
data.frame(
size = c(sizes, sizes),
time = c(runtimes, runtimes2),
method = c(rep("epistack", 8), rep("EnrichedHeatmap", 8))) |>
ggplot(aes(x = size, y = time, color = method)) +
geom_line() +
geom_point(shape = 3) +
theme_bw(base_size = 14) +
labs(x = "Number of regions", y = "Time (s)",
title = "plotEpistack() benchmark") +
coord_cartesian(ylim = c(0, 60))
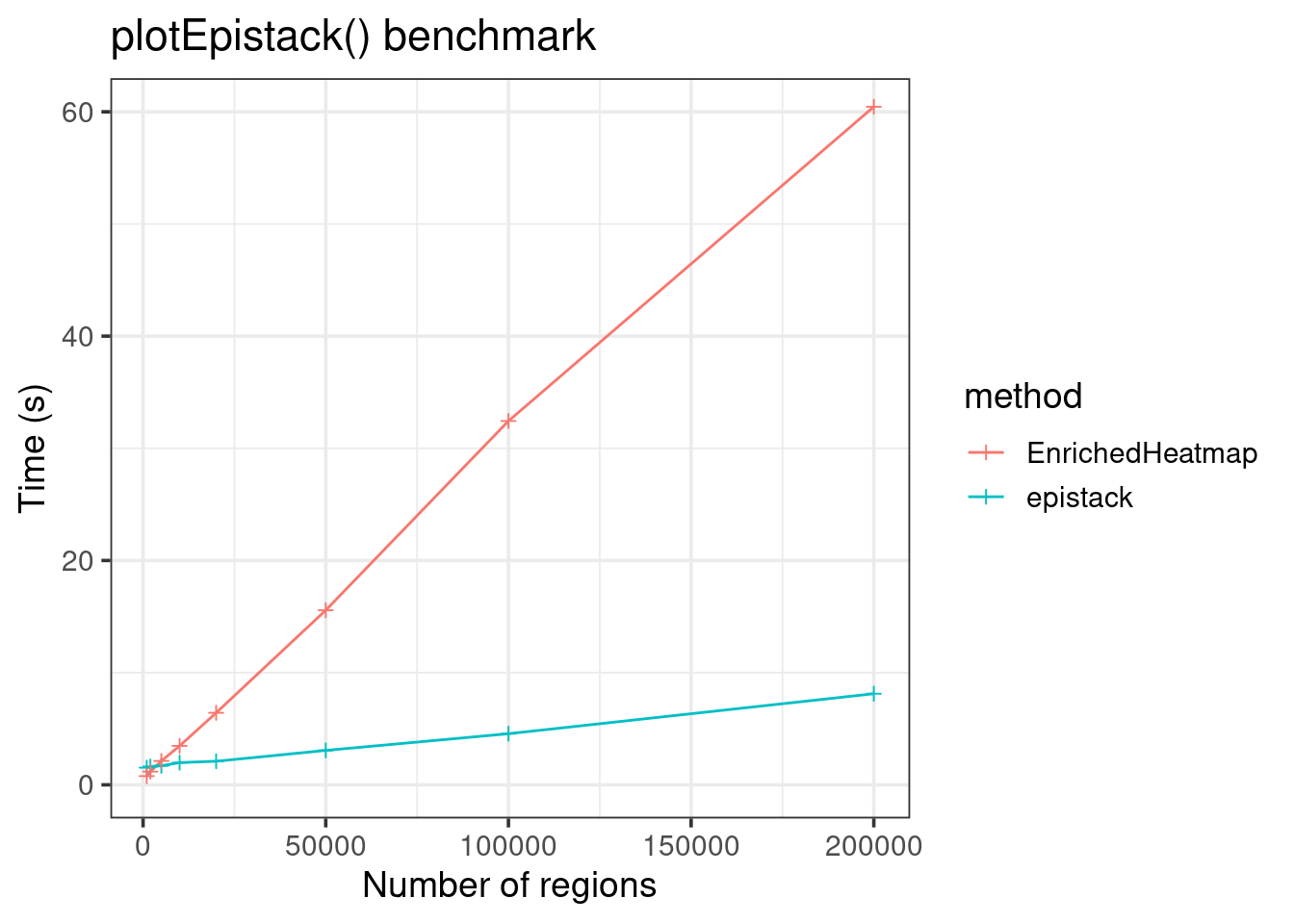
EnrichedHeatmp is less fast, but can still plot up to hundred thousands of regions!
Making plotEpistack outputs similar to EnrichedHeatmap outputs
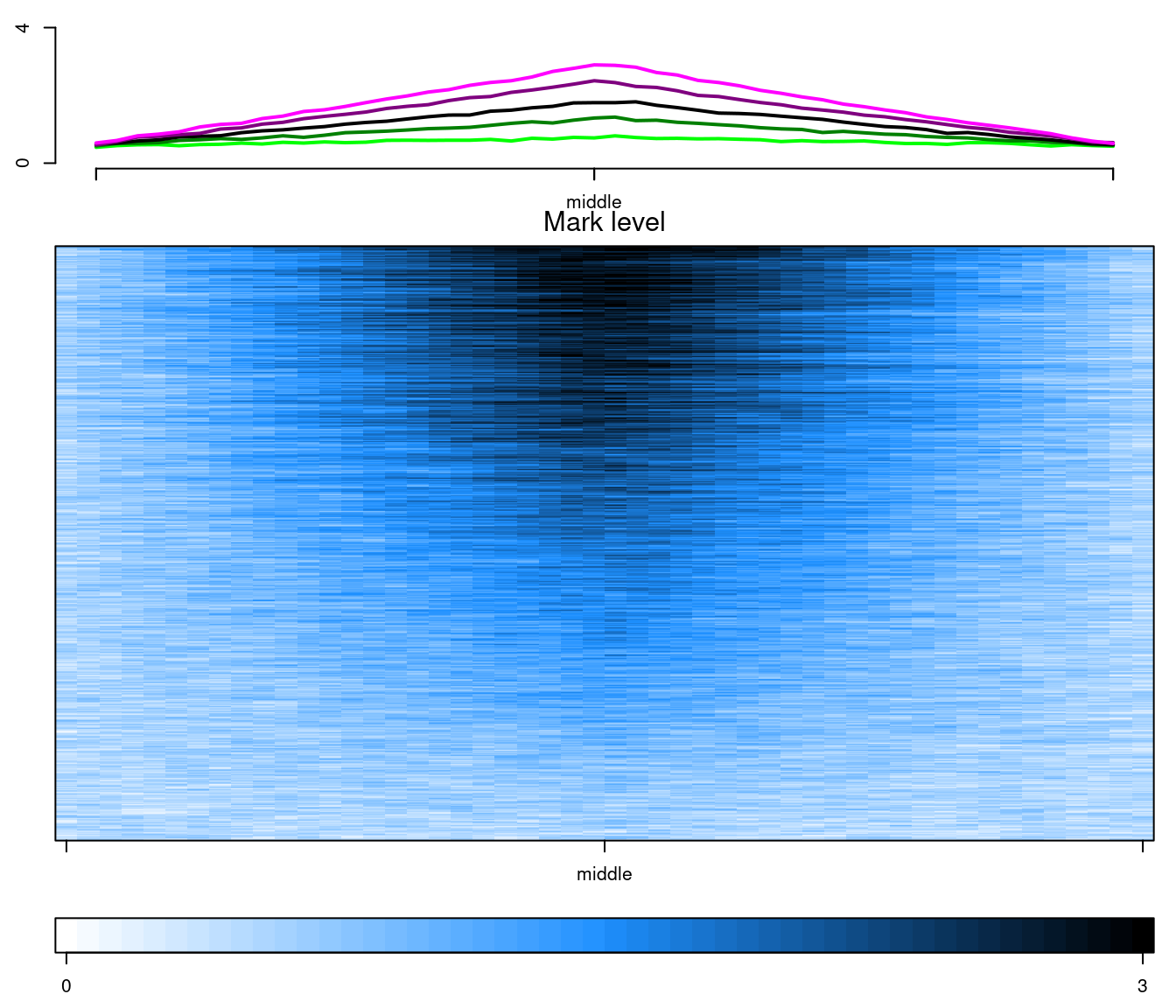
In the previous benchmark, we used the amazing flexibility of EnrichedHeatmap to make its outputs similiar to epistack outputs. It is alsa possible to do the opposti: make epistack output more similar to the EnrichedHeatmap outputs, as illustrated bellow:
testdata <- benchdata[[1]]
testdata$bins <- NULL
layout(matrix(1:3, ncol = 1), heights = c(1, 3, 0.5))
old_par <- par(mar = c(2.5, 2.5, 1, 1))
plotAverageProfile(
testdata, pattern = "mcols",
x_labels = c("", "middle", ""), ylim = c(0, 4)
)
plotStackProfile(
testdata, pattern = "mcols",
x_labels = c("", "middle", ""),
zlim = c(0, 3), palette = colorRampPalette(c("white", "dodgerblue", "black")),
title = "Mark level"
)
plotStackProfileLegend(
zlim = c(0, 3),
palette = colorRampPalette(c("white", "dodgerblue", "black"))
)
par(old_par)
layout(1)
Session Info
sessionInfo()
#> R version 4.1.1 (2021-08-10)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
#> LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
#>
#> locale:
#> [1] LC_CTYPE=fr_FR.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=fr_FR.UTF-8 LC_COLLATE=fr_FR.UTF-8
#> [5] LC_MONETARY=fr_FR.UTF-8 LC_MESSAGES=fr_FR.UTF-8
#> [7] LC_PAPER=fr_FR.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=fr_FR.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] grid parallel stats4 stats graphics grDevices utils
#> [8] datasets methods base
#>
#> other attached packages:
#> [1] EnrichedHeatmap_1.22.0 ComplexHeatmap_2.9.4 ggplot2_3.3.5
#> [4] epistack_0.99.0 GenomicRanges_1.44.0 GenomeInfoDb_1.28.1
#> [7] IRanges_2.26.0 S4Vectors_0.30.0 BiocGenerics_0.38.0
#>
#> loaded via a namespace (and not attached):
#> [1] locfit_1.5-9.4 Rcpp_1.0.7 lattice_0.20-45
#> [4] circlize_0.4.13 png_0.1-7 assertthat_0.2.1
#> [7] digest_0.6.28 foreach_1.5.1 utf8_1.2.2
#> [10] R6_2.5.1 evaluate_0.14 highr_0.9
#> [13] blogdown_1.5 pillar_1.6.2 GlobalOptions_0.1.2
#> [16] zlibbioc_1.38.0 rlang_0.4.11 jquerylib_0.1.4
#> [19] magick_2.7.3 GetoptLong_1.0.5 rmarkdown_2.11
#> [22] labeling_0.4.2 stringr_1.4.0 RCurl_1.98-1.3
#> [25] munsell_0.5.0 compiler_4.1.1 xfun_0.26
#> [28] pkgconfig_2.0.3 shape_1.4.6 htmltools_0.5.2
#> [31] tidyselect_1.1.1 tibble_3.1.5 GenomeInfoDbData_1.2.6
#> [34] bookdown_0.22 codetools_0.2-18 matrixStats_0.60.1
#> [37] fansi_0.5.0 viridisLite_0.4.0 crayon_1.4.1
#> [40] dplyr_1.0.7 withr_2.4.2 bitops_1.0-7
#> [43] jsonlite_1.7.2 gtable_0.3.0 lifecycle_1.0.1
#> [46] DBI_1.1.1 magrittr_2.0.1 scales_1.1.1
#> [49] stringi_1.7.5 farver_2.1.0 XVector_0.32.0
#> [52] doParallel_1.0.16 bslib_0.3.1 ellipsis_0.3.2
#> [55] generics_0.1.0 vctrs_0.3.8 RColorBrewer_1.1-2
#> [58] rjson_0.2.20 iterators_1.0.13 tools_4.1.1
#> [61] glue_1.4.2 purrr_0.3.4 plotrix_3.8-1
#> [64] fastmap_1.1.0 yaml_2.2.1 clue_0.3-59
#> [67] colorspace_2.0-2 cluster_2.1.2 knitr_1.36
#> [70] sass_0.4.0
Guillaume Devailly
Researcher in farm animal epigenetics
Researcher in farm animal epigenetics at INRAE, France.